🌀 Fluorescence Polarization / Anisotropy (FP/FA)
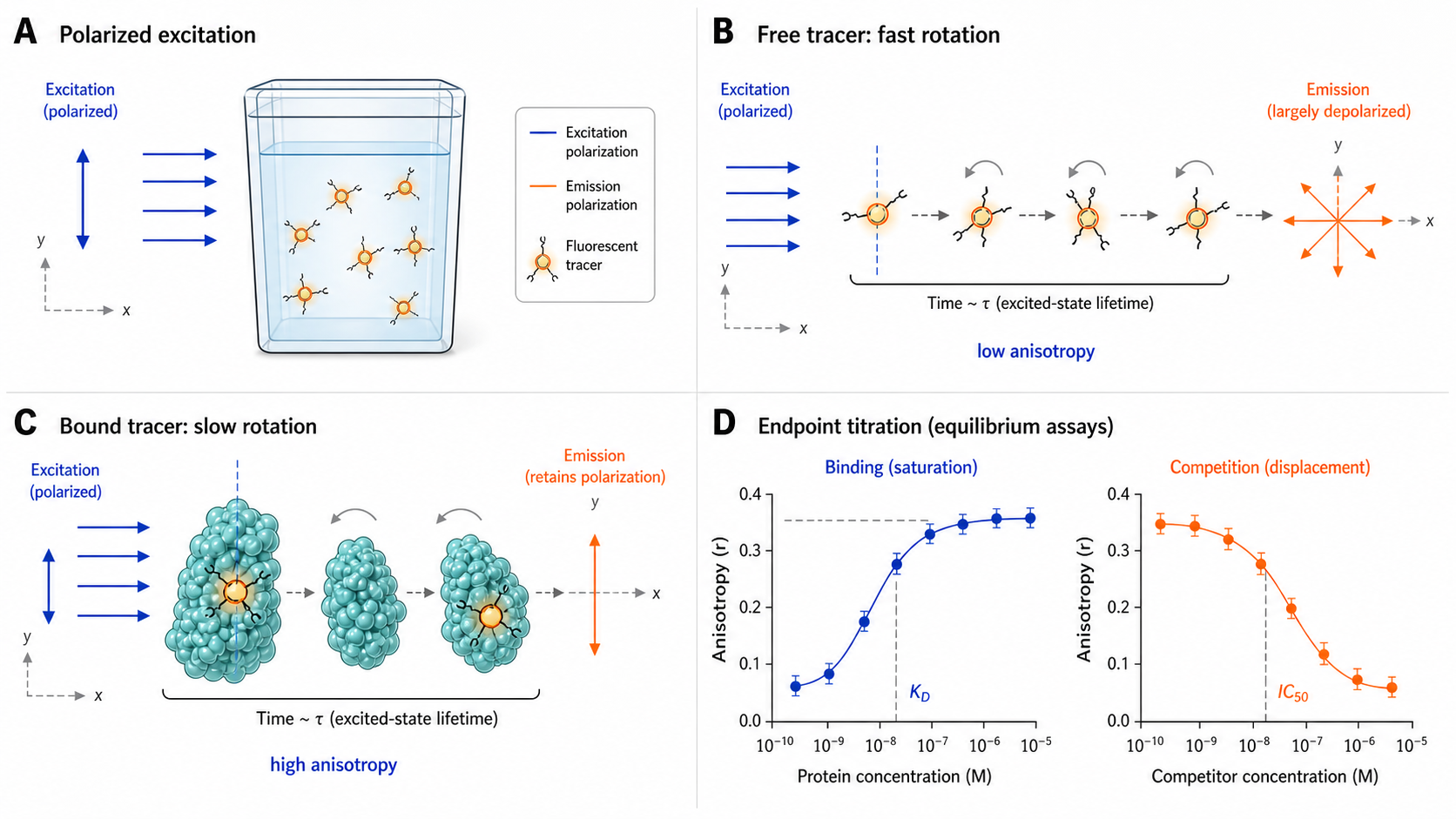
Fluorescence polarization measures molecular rotation. When you excite a fluorophore with polarized light, the emitted light remains polarized only if the molecule hasn't tumbled during the fluorescence lifetime. A small, free tracer tumbles fast → low polarization. The same tracer bound to a large protein complex tumbles slowly → high polarization. Plot polarization vs. ligand concentration → sigmoidal binding curve → KD. In drug screening, the most common format is competition FP, where a labeled tracer is pre-bound to the target and unlabeled compounds compete for the binding site — yielding Ki values for entire compound libraries.
FP is the workhorse of high-throughput screening (HTS) in drug discovery. Mix-and-read, no wash steps, no separation, no immobilization, compatible with 384- and 1536-well plates. It's been a pharma staple since the 1990s for good reason: it's simple, robust, and scalable. While this page uses "protein" as the default binding partner for readability, FP works for any macromolecule — DNA, RNA, lipid vesicles, carbohydrates — as long as the MW ratio is sufficient.
FP measures equilibrium KD (like MST and ITC) — it does not give kinetic rate constants (ka, kd). For kinetics, use SPR or BLI. Unlike MST, FP requires a fluorescent tracer — either a fluorescently labeled ligand or a naturally fluorescent compound. The key constraint: the molecular weight ratio between the bound complex and the free tracer (MWcomplex / MWtracer) must be large enough to produce a measurable change in rotational correlation time — typically at least 5× in MW (conservative assays use 10×).
Key Physics Concepts
Polarized Excitation & Emission
When plane-polarized light excites a fluorophore, only molecules whose absorption transition dipole is aligned with the electric field vector absorb efficiently — photoselection. If the molecule doesn't rotate before emitting, the emission retains its polarization. If it rotates during the fluorescence lifetime τ, the emission is depolarized.
Polarization (mP) and Anisotropy (r)
Two related observables from the same measurement:
Anisotropy adds linearly for mixtures: robs = fbound·rbound + (1 − fbound)·rfree. Polarization does not — always fit in r, not mP.
The Perrin Equation
r = r₀ / (1 + τ/φ)r₀ = limiting anisotropy (≤ 0.4), τ = fluorescence lifetime (ns), φ = rotational correlation time (ns). Rule of thumb: φ (ns) ≈ MW (kDa) × 0.4 at 20°C in water. When τ ≈ φ: maximum sensitivity to size changes.
Interactive FP Simulator
Explore the physics of fluorescence polarization through interactive simulations.
Parameters
How FP Works — Measurement Principle
🔭 The Optical Setup
- Excitation light passes through a polarizing filter (vertical), so only vertically polarized light hits the sample.
- Emission is split by a polarizing beam splitter into I‖ (parallel) and I⊥ (perpendicular) channels.
- Both intensities are measured simultaneously by two PMTs (T-format) or sequentially by rotating a polarizer (L-format). T-format is standard in plate readers.
- r = (IVV − G·IVH) / (IVV + 2G·IVH), where G corrects for differential detector sensitivity.
⚖️ The G-Factor
The G-factor corrects for differential sensitivity of the detection system to horizontally vs. vertically polarized emission:
G = I_HV / I_HHH excitation, V emission / H excitation, H emission
Without G-factor correction, systematic errors of 10–20% in r are common. Typical G values: 0.9–1.1. Modern plate readers auto-measure G with a free-dye sample — always calibrate.
Segmental Motion — The Hidden Assay Killer
The Perrin equation assumes the fluorophore is rigidly attached to the macromolecule. In reality, a dye connected via a flexible linker undergoes local rotation (segmental motion) independent of the overall protein tumbling. The correct steady-state expression (Lipari-Szabo model-free):
r = r₀ × [S² × φglobal/(φglobal + τ) + (1 − S²) × φeff/(φeff + τ)]1/φeff = 1/φlocal + 1/φglobal — both motions contribute to depolarization
S² = order parameter (0–1)
S² = 1: rigid attachment → reduces to Perrin equation
S² → 0: dye wobbles freely → r dominated by fast φ_eff term
S² ≈ 0.7–0.9 for direct NHS lysine labeling (short linker)
S² ≈ 0.1–0.3 for dye on PEG₄ tether (flexible linker)
Example — 50 kDa protein:
φ_global = 20 ns, τ = 4 ns (FITC), flexible linker (φ_local = 0.3 ns, S² = 0.3)
Perrin prediction (rigid): r = 0.333
Lipari-Szabo (flexible): r = 0.119
Flexible linker cuts r_bound by 64%!
Fix: Keep the linker between dye and binding moiety short and rigid when possible. If r_bound is disappointingly low, try a different labeling position or shorter linker before changing fluorophores.
Assay Design — Key Experimental Parameters
FP Assay Design Checklist
- Define the binding pair — What binds what? Which component gets labeled?
- Choose the tracer — Label the smaller binding partner for maximum Δr.
- Select the fluorophore — Match τ to the MW range (see Perrin Explorer). Fluorescein is the default.
- Validate the tracer — Measure KD of labeled tracer by direct saturation binding. Compare to unlabeled KD.
- Optimize [Tracer] — Use the lowest concentration that gives acceptable S/N. Rule: [T] < KD/10 for accurate KD.
- Run saturation binding — Titrate protein vs. fixed [tracer] → determine KD,tracer and confirm r_free and r_bound.
- Set up competition — Fix [protein] and [tracer]; [P] should be near KD,tracer for optimal sensitivity.
- Equilibration time — Run a time course (0, 15, 30, 60, 90 min). Typical: 15–60 min at RT.
- Controls — Tracer-only (baseline), tracer + saturating protein (max), DMSO blanks, positive control compound.
- Plate layout — Randomize or interleave; avoid edge effects.
Tracer Concentration Regime
| [T] vs KD | Status | Consequence |
|---|---|---|
| [T] ≤ KD/10 | ✅ Ideal | EC₅₀ ≈ KD. Simple binding model valid. |
| KD/10 < [T] < KD | ⚠️ Use quadratic | EC₅₀ shifts right. Use quadratic model; report [T]. |
| [T] >> KD | ❌ Danger | Measuring stoichiometry. KD is an upper limit. Reduce [T]. |
Protein Concentration in Competition
| [Protein] vs KD,tracer | Quality | Reason |
|---|---|---|
| [P] = KD,tracer | ✅ Optimal | 50% of tracer is bound → maximum dynamic range for displacement |
| [P] = 3–5× KD,tracer | ⚠️ Acceptable | >75% tracer bound → still workable; need more competitor to displace |
| [P] >> KD,tracer | ❌ Problem | Nearly all tracer bound → competitors need extreme concentrations → lose sensitivity to weak binders |
Assay Quality: Z′ Factor
Z′ = 1 − (3σpos + 3σneg) / |µpos − µneg|| Z′ | Quality |
|---|---|
| > 0.5 | ✅ Excellent — suitable for HTS |
| 0.2 – 0.5 | ⚠️ Marginal — optimize before screening |
| < 0.2 | ❌ Unacceptable — redesign the assay |
Fluorophore Selection Guide
Fluorescein (FITC)
λ_ex/λ_em: 494/521 nm · τ ≈ 4.0 ns
✓ Universal, cheap, well-characterized
✗ pH-sensitive (pKa ≈ 6.4) — use buffer pH ≥ 7.5 for stable FP; photobleaches
Best for: small molecule tracers (0.3–2 kDa) binding to proteins 10–500 kDa
TAMRA
λ_ex/λ_em: 555/580 nm · τ ≈ 4.2 ns
✓ pH-insensitive, photostable, red-shifted
✗ More expensive, hydrophobic
Best for: assays where fluorescein gives problems; peptide tracers
Alexa Fluor 488
λ_ex/λ_em: 495/519 nm · τ ≈ 4.1 ns
✓ Brighter & more stable than FITC; pH-insensitive
✗ More expensive
Best for: high-quality assays where stability matters
BODIPY-FL
λ_ex/λ_em: 503/512 nm · τ ≈ 5.7 ns
✓ Narrow emission, high QY, pH-insensitive
✗ Hydrophobic — can cause non-specific binding
Best for: when spectral purity is important
Dansyl
λ_ex/λ_em: 335/518 nm · τ ≈ 10–15 ns
✓ Longest conventional τ — ideal for peptide tracers (2–20 kDa)
✗ UV excitation; τ is environment-dependent (changes upon binding!)
⚠ Verify τ stability before committing to quantitative KD extraction
Best for: peptide/small protein tracers binding large complexes
Cy5 / Alexa Fluor 647
λ_ex/λ_em: 649/670 nm · τ ≈ 1.0 ns
✓ Far-red, minimal autofluorescence
✗ Short τ compresses window for peptide-sized tracers
Best for: small-molecule tracers in autofluorescent samples
Fluorophore Decision Tree
Is your tracer a small molecule (<2 kDa)?
→ YES: Fluorescein or Alexa 488 (τ ≈ 4 ns = ideal)
→ NO: Is it a peptide (2–20 kDa)?
→ YES: TAMRA, Alexa 488, or dansyl (longer τ for bigger peptides)
→ NO: Is it a protein?
→ Is the binding partner >5× larger?
→ YES: Any dye works; fluorescein is cheapest
→ NO: Use long-lifetime dyes (pyrene) or TR-FRET
Is autofluorescence a problem (cell lysates, serum)?
→ YES: Alexa 647 / Cy5 or TR-FP (time-gated)
Are you doing HTS (>10,000 wells)?
→ YES: Fluorescein (cheapest, well-validated)
Common FP Assay Formats
Saturation Binding
Fix [tracer], titrate [protein]. Anisotropy increases as tracer binds → saturation curve → KD,tracer, r_free, r_bound. Always run this first to characterize the tracer before competition assays.
Competition / Displacement
Fix [protein] + [tracer] at equilibrium; titrate unlabeled competitor. Anisotropy decreases → IC₅₀ → Ki via Cheng-Prusoff. Most common format in drug discovery — screen compound libraries.
Enzyme-Linked FP
Fluorescently labeled substrate; enzyme cleaves it → smaller product → anisotropy drops. Inhibitors prevent cleavage → high anisotropy. Yields IC₅₀ of inhibitor. Used for protease, kinase, nuclease assays. Note: functional assay, not direct binding.
Ternary Complex (PROTACs)
Label one protein; add bifunctional compound + second protein. Ternary complex formation → MW increase → higher anisotropy. Hook effect: at high [compound], bell-shaped dose-response. EC₅₀ of rising phase reflects potency; hook concentration = binary saturation.
Data Analysis
Binding Models
Intensity weighting: Anisotropy is additive over intensity fractions. If quantum yield changes upon binding, use the intensity-weighted model. Always check total fluorescence intensity across the titration — if it changes >15–20%, use the intensity-weighted model or switch fluorophores.
Direct Saturation (simple)
r = r_free + (r_bound − r_free) × [P] / ([P] + KD)Valid when [T] << KD
Saturation (quadratic — with tracer depletion)
[PT] = ½{([P]_t + [T]_t + KD) − √(…² − 4[P]_t[T]_t)}
r = r_free + (r_bound − r_free) × [PT] / [T]_tCompetition (Cheng-Prusoff approximation)
Ki = IC₅₀ / (1 + [T]free / KD,tracer)[T]_free ≈ [T]_total when fraction bound is small
Important: r_top ≠ r_bound
In competition, r_top (starting anisotropy) depends on fraction bound at the chosen [P] and KD,tracer. At [P] ≈ KD,tracer, only ~50% is bound → r_top ≈ (r_free + r_bound)/2. Use measured control values, not theoretical ones.
Common Pitfalls in FP Data Analysis
Confusing EC₅₀ with K_D
EC₅₀ = K_D only when [tracer] << K_D. Otherwise EC₅₀ > K_D. Always report which one you measured.
Fitting in mP instead of r
Polarization does not add linearly for mixtures. Best practice: convert to r, fit, convert back to mP for display.
Neglecting the G-factor
Without instrument correction, r is systematically wrong. Run a free-dye calibration every session.
Inner filter effect
High [fluorophore] or compound absorbs excitation/emission light. Keep OD < 0.1 at both λ_ex and λ_em.
Compound autofluorescence / quenching
Compounds may fluoresce (especially green channel!) or quench the tracer. Run a tracer-only + compound control.
Temperature sensitivity
Viscosity drops ~2–3% per °C → φ decreases → r decreases. A 5°C drift across a plate causes systematic error.
Aggregation / promiscuous binding
Colloidal aggregates sequester protein → apparent displacement. Add 0.01–0.05% Triton X-100 to discriminate.
DMSO mismatch
DMSO increases viscosity → r increases. Keep DMSO constant across all wells including controls.
Light scattering at high [protein]
At high protein (>10 µM), Rayleigh scattering increases apparent polarization. Add long-pass emission filter.
PMT saturation
At very high fluorescence, nonlinear detector response distorts the I‖/I⊥ ratio. Keep counts in the linear range.
Viscosity modulation — deliberate and accidental
Adding glycerol or sucrose slows tumbling and increases r_bound — a useful lever for assays where Δr is too small. The flip side: viscosity changes with temperature, freeze-thaw history, and buffer batch; even 5% glycerol variation shifts r. Keep buffer composition identical across all wells, including blanks and controls.
Strengths & Limitations
| ✅ Strengths | ❌ Limitations |
|---|---|
| Homogeneous — mix-and-read, no wash steps | Requires fluorescent tracer (labeled or intrinsic) |
| HTS-compatible: 384-well and 1536-well | MW ratio must be ≥5× for adequate Δr |
| Equilibrium K_D in solution | Cannot distinguish competitive vs. allosteric binding |
| Fast — minutes to equilibrium, seconds to read | Fluorescence interference: autofluorescent/colored compounds |
| Very low sample: µL volumes, nM concentrations | No kinetic information (k_a, k_d) |
| Simple instrumentation (any FP plate reader) | Inner filter effect at high concentrations |
| No immobilization — true solution measurement | Temperature-sensitive (viscosity changes affect r) |
| Robust — validated protocols for hundreds of targets | Protein–protein interactions (both large) give minimal Δr |
| Competition format gives K_i for unlabeled compounds | Aggregating/promiscuous compounds give false positives |
| TR-FP variants reduce background for crude samples | Cheng-Prusoff K_i is approximate; exact solutions are complex |
Time-Resolved Fluorescence Polarization (TR-FP)
TR-FP uses a time gate to reject short-lived background before measuring FP. The canonical implementations use long-lifetime organic fluorophores — Ru(II) polypyridyl complexes (τ ≈ 100–1000 ns), pyrene (τ ≈ 100–400 ns in deoxygenated solvent), acridone, dansyl — or fluorescence-lifetime gating on conventional dyes. After pulsed excitation, wait long enough for autofluorescence (ns lifetime), scattered excitation, and plate background to decay before integrating emission.
Note on lanthanides: lanthanide chelates (Eu³⁺, Tb³⁺, τ ≈ 100–1000 µs) are sometimes loosely grouped under "TR-FP," but their f–f emission has a poorly defined emission dipole and near-zero intrinsic anisotropy (r₀ ≈ 0). They are the basis of TR-FRET assays (LanthaScreen, HTRF), not of practical TR-FP — even with a perfect time gate, there is no polarization signal to measure.
Common misconception: TR-FP does NOT extend the MW sensitivity range
Even at the long end of organic-dye lifetimes, τ has to be matched to φ for a measurable r. Hypothetically with a τ ≈ 500 µs lanthanide and a 500 kDa complex (φ ≈ 200 ns), τ/φ ≈ 2500 → r ≈ 0 — fully depolarized (and lanthanide r₀ is already ≈ 0). The time gate eliminates background; it does not shift the sensitive MW window. To extend that window you must change the fluorophore τ, not gate on it.
TR-FP excels at:
- HTS in crude matrices (cell lysates, serum, conditioned media)
- Colored compound libraries — autofluorescence gone after time gate
- Higher Z′ values — reduced background variance
To extend FP to larger complexes, use:
- Pyrene (τ ≈ 100–400 ns) → matches φ of 250–1000 kDa complexes
- Acridone (τ ≈ 15–30 ns) → matches φ of 40–75 kDa complexes
- Dansyl (τ ≈ 10–15 ns) → matches φ of 25–40 kDa complexes
TR-FP vs TR-FRET: TR-FP measures polarization of donor emission only — same binding events as conventional FP, lower background. TR-FRET measures sensitized acceptor emission after energy transfer — proximity-based, requires labeling both partners, more common in HTS due to dual-wavelength specificity.
When NOT to Use FP
❌ When both binding partners are large
A 50 kDa protein binding a 60 kDa protein: MW changes from 50 → 110 kDa. With fluorescein (τ = 4 ns): r_free ≈ 0.333, r_bound ≈ 0.367, Δr = 0.034 — too small. Use SPR, MST, or ITC instead.
❌ When you need kinetics
FP is equilibrium only. No k_a, k_d, or residence time information. Use SPR or BLI for kinetics.
❌ When binding changes fluorophore properties
If binding quenches or enhances fluorescence >20%, the intensity-weighted average is unreliable. Check total fluorescence across the titration.
❌ When compound autofluorescence dominates
Some chemical libraries have significant green fluorescence. Screen in the red channel (TAMRA, Cy5) or use time-resolved FP.
❌ For fragment screening
Fragment K_D is typically 0.1–10 mM — requires mM concentrations, causing inner filter effects and solubility issues. Better: TSA/DSF, SPR, or ligand-observed NMR (STD, WaterLOGSY).
❌ When label-free is required
FP requires a fluorophore. If modification is not acceptable, use SPR, BLI, or MST label-free (UV detection of tryptophan).
FP for Drug Discovery
🔍 Primary Screening (HTS)
- 384/1536-well competition format
- Screen 10,000–1,000,000 compounds at single concentration
- Z′ > 0.5 required
- Hits: Δr > 3σ below control mean
- Cost: ~$0.05–0.20 per well (reagent cost)
- Throughput: 50,000–100,000 data points/day/reader
✅ Hit Validation & Dose-Response
- Confirm hits with 8–12 point dose-response (half-log dilution)
- Determine IC₅₀ for each confirmed hit
- Convert IC₅₀ → Ki with Cheng-Prusoff
- Counter-screens: autofluorescence, aggregation (Triton test)
- Orthogonal validation: SPR, MST, or ITC for top compounds
⚗️ SAR & Lead Optimization
- 16–24 analogs per plate, full dose-response each
- Compare Ki values to guide medicinal chemistry
- Monitor selectivity vs. off-target proteins in parallel
- Feeds into biophysics cascade alongside SPR characterization
FP vs Other Techniques
🌀 FP/FA
- Equilibrium KD (via Ki in competition)
- Requires fluorescent tracer
- HTS-compatible (384/1536-well)
- µL volumes, nM concentrations
- Fastest readout (seconds per well)
- MW ratio constraint
- No kinetics, no thermodynamics
🔬 MST
- Equilibrium KD
- Requires fluorescent label (or label-free UV)
- 16-point capillary format
- No MW constraint (works for any size)
- Works in crude lysates
- Lower throughput than FP for HTS
- No kinetics, no thermodynamics
✨ SPR
- Kinetics (ka, kd, KD)
- Label-free
- Requires surface immobilization
- Gold standard for drug–target kinetics
- Higher throughput with multi-channel systems
- Regulatory acceptance for biosimilars
- Expensive instruments
Key message: FP excels at high-throughput equilibrium screening. MST excels at solution-phase characterization without MW constraints. SPR excels at kinetic characterization. They are complementary, not competitive.
Publication Checklist
Experimental Design
- ☐Tracer K_D determined by direct saturation binding
- ☐[Tracer] stated and < K_D/10 (or justified)
- ☐[Protein] in competition ≈ K_D,tracer (or justified)
- ☐G-factor measured and applied
- ☐Temperature controlled and reported
- ☐Equilibration time validated (time course)
- ☐DMSO concentration matched across all wells
Data Quality
- ☐Z′ > 0.5 for screening data
- ☐Δr (or ΔmP) reported
- ☐Both baselines (free and bound) well-defined
- ☐Counter-screen for compound interference
- ☐≥ 3 independent replicates for K_D/K_i values
- ☐Total fluorescence intensity checked (no quenching)
- ☐r_bound compared to Perrin prediction — segmental motion noted if low
Reporting
- ☐K_D or K_i ± CI (not just IC₅₀)
- ☐Binding model specified (Hill, simple, quadratic)
- ☐Cheng-Prusoff correction applied and stated
- ☐Fluorophore, wavelengths, and reader model reported
- ☐Raw anisotropy or mP data shown (not just normalized)
- ☐Orthogonal validation for key compounds (SPR, ITC, MST)
Instruments using Fluorescence Polarization
- BMG LABTECH PHERAstar FSX — gold standard FP plate reader, dual PMT (T-format), TR-FP module
- BMG LABTECH CLARIOstar Plus — excellent FP module with LVF monochromators
- Tecan Spark — multimode reader with FP capability
- Tecan Infinite M1000 Pro — quad-monochromator, good FP performance
- PerkinElmer EnVision — HTS workhorse, TRF and FP modules, TR-FP
- Molecular Devices SpectraMax — FP-capable multimode readers
- BioTek Synergy Neo2 — dual PMT, good FP with temperature control
Have SPR or BLI data?
Upload your raw files and get an automated kinetic analysis in minutes. We support Biacore, Octet, and other major formats.
Upload & Analyze