🔬 Microscale Thermophoresis (MST)
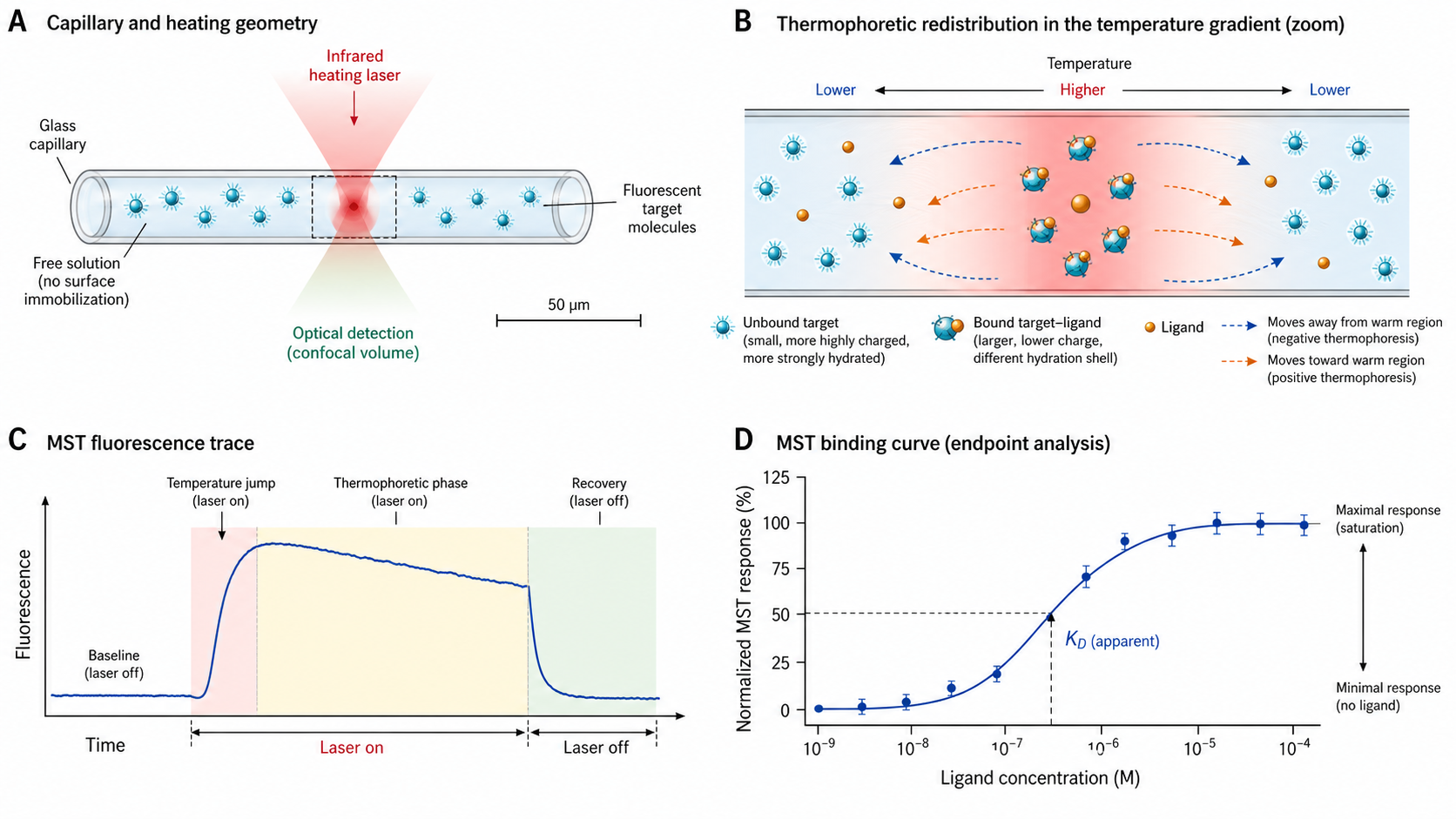
MST is one of the most versatile binding assays in biophysics: no surface, no size limit, works in crude lysates, and needs just microliters of sample. It measures molecular interactions in solution by detecting changes in the thermophoretic movement of fluorescently labeled molecules along a microscopic temperature gradient. When a ligand binds to the labeled target, it alters the target's size, charge, hydration shell, and conformation — all of which affect how the molecule moves in a temperature gradient. This change is detected as a shift in fluorescence distribution, yielding a dose-response curve from which the dissociation constant (KD) is determined. No immobilization, no mass limitation — MST works in true solution with as little as ~4 µL of sample per capillary.
Key Physics Concepts
Thermophoresis (Soret Effect)
Directed movement of molecules along a temperature gradient (Ludwig, 1856; Soret, 1879; modern MST foundations: Duhr & Braun, PNAS 2006; Wienken et al., Nat Commun 2010). Molecules typically move from hot to cold regions (positive Soret coefficient ST), but ST can also be negative under certain salt/temperature conditions (Iacopini & Piazza 2003), so the sign of ΔFnorm is not predictable a priori. ST depends on molecular size, charge, hydration shell, and conformation — binding a ligand changes one or more of these, producing a measurable signal.
Binding-Induced Signal Change
When a ligand binds, the complex has different thermophoretic properties. Four contributors: size (complex is larger), charge (altered surface charge distribution), hydration shell (often the dominant factor — binding buries or exposes polar residues), and conformation (structural rearrangements). The weighted average of bound and unbound states produces a measurable shift in Fnorm.
Dose-Response Quantification
Titrate increasing concentrations of unlabeled ligand against a constant concentration of labeled target. Each concentration gives one Fnorm value. Plot Fnorm vs [Ligand] → sigmoidal dose-response curve → fit to binding isotherm → KD. This is an equilibrium measurement — MST gives KD but not kinetic rate constants (ka, kd).
Interactive MST Simulator
Explore microscale thermophoresis: fluorescence traces, dose-response curves, and experiment planning
💡 Why do bound complexes move differently?
The Soret coefficient (ST) determines how strongly a molecule is pushed by a temperature gradient. It depends on the molecule's size, charge, hydration shell, and conformation. When a ligand binds to the target, all four properties change — the complex is larger, its surface charge distribution shifts, and water molecules rearrange around it. In most cases, this increases ST, causing the bound complex (orange) to deplete faster from the heated zone than the unbound target (cyan). However, the sign and magnitude of the change are empirical — some interactions decrease ST or alter only the T-jump component. This differential behavior is what MST measures — the fluorescence change in the detection zone differs depending on how much target is bound.
MST Signal Phases
A single MST trace records fluorescence over ~40 seconds. Understanding the three distinct phases is essential for correct data analysis — and for distinguishing MST from related measurement modes (TRIC, Spectral Shift).
⚡ T-Jump (<1 s)
Rapid fluorescence change immediately after IR laser turns on. This is not thermophoresis — it reflects the temperature-related intensity change (TRIC) of the fluorophore: dominantly a quantum-yield shift, but modulated by the dye's local hydration shell and electronic environment, which is precisely why TRIC reports binding. Happens in <1 s. Some analyses use this phase alone (the "TRIC" evaluation).
🔬 Thermophoresis (5–30 s)
Gradual fluorescence change as molecules redistribute in the temperature gradient. This is the actual MST signal. Exponential approach to a steady state where thermophoretic drift and back-diffusion balance. Time constant τMST ≈ 5–15 s, depending on molecular size.
↩️ Back-Diffusion (5–20 s)
After the IR laser turns off, molecules diffuse back to a uniform distribution. Time constant τback ≈ 5–20 s (governed by Fick's law and molecular diffusion coefficient — larger molecules diffuse back more slowly). Not to be confused with thermal relaxation (~1–3 s).
🔑 MST vs TRIC vs Spectral Shift — Three Distinct Measurements
How MST Works
The Measurement Principle
A focused IR laser (1480 nm, absorbed by water) creates a microscopic temperature gradient inside a glass capillary (~100–200 µm inner diameter). The heated spot is ~50–100 µm FWHM, producing a local temperature increase of 2–6 K. Labeled target molecules redistribute in this gradient (thermophoresis). Fluorescence is excited by an LED and measured by a detector — the ratio of fluorescence after thermophoresis to before gives Fnorm.
From Traces to Binding Curve
Each capillary contains the same concentration of labeled target + a different concentration of unlabeled ligand. Typically 16 capillaries with 1:1 serial dilution. Each capillary produces one MST trace → one Fnorm value. Plotting Fnorm vs [Ligand] yields the dose-response curve, fitted to a 1:1 binding model (or Hill equation) to extract KD. Signal-to-noise ratio (response amplitude / noise) determines measurement quality — SNR > 5 is the minimum threshold.
Labeling Strategies
MST requires a fluorescent signal. Choose your labeling approach based on your target protein and experimental goals. The decision tree below the cards helps you pick the right one.
Fluorescent Protein Fusions
GFP, EGFP, mCherry, YFP fused genetically to the target. Site-specific, homogeneous. Requires cloning and expression; tag may affect function.
Best for: Recombinant proteins already tagged. LED: Blue (470 nm) or green channel.
Chemical Labeling (NHS-ester)
NHS esters react with primary amines (lysine ε-amino groups). NanoTemper RED-NHS, Alexa Fluor 647, Cy5-NHS all work. Target degree of labeling (DOL) 0.5–1.0.
Best for: Any purified protein without His-tag. LED: Red (625–650 nm).
His-tag Labeling (RED-tris-NTA)
Tris-NTA chelator binds 6×His-tag with low-nM affinity (typically ~1–10 nM, depending on tag length and Ni²⁺ loading). One-step, no purification needed. Works in crude lysates. Keep imidazole < 5–10 mM; no EDTA.
Best for: Rapid screening, crude lysates, His-tagged proteins.
Maleimide (Thiol-Reactive)
Reacts with cysteine sulfhydryl groups. More site-specific than NHS-ester labeling. Ideal when you have an accessible, non-essential cysteine or an engineered one.
Best for: Site-specific labeling when cysteine is available.
SNAP-tag / HaloTag
Self-labeling protein tags with fluorescent substrates. Very site-specific, highly efficient. Requires genetic fusion but offers excellent control over label position.
Best for: Precise site-specific labeling; growing in popularity.
Intrinsic Fluorescence (Label-Free)
Uses intrinsic tryptophan fluorescence (excitation ~280 nm). Truly label-free — no modification. Requires Trp residues near the binding site. Lower sensitivity, higher protein concentrations needed (100 nM–10 µM). Requires UV optics (NT.LabelFree).
Best for: Membrane proteins, native protein validation.
🌳 Choosing Your Labeling Strategy
Is your protein His-tagged?
→ YES: RED-tris-NTA (fastest, works in lysate)
→ NO: Is it already a fluorescent fusion (GFP, mCherry)?
→ YES: Use the FP channel
→ NO: Is purified protein available?
→ YES, accessible Cys: Maleimide dye
→ YES, no specific Cys: NHS-ester dye (RED-NHS)
→ NO: Can you add a tag? → SNAP/HaloTag or His-tag
→ NO modification possible: Intrinsic Trp fluorescence (UV)
Experimental Design — Step-by-Step Protocol
10-Step MST Protocol
- Choose labeling strategy — use the decision tree above
- Label your target — follow the protocol for your chosen approach; verify activity post-labeling
- Determine optimal [Target] — run an LED scan; aim for 200–2000 fluorescence counts; use ≤ KD/10 if possible
- Prepare serial dilution — 16 points, 1:1 dilution; range from ~20× above to ~20× below expected KD
- Mix target + ligand — combine equal volumes; allow equilibration (10–15 min at RT)
- Load capillaries — standard treated (default), premium coated (sticky proteins), or hydrophobic (membrane proteins)
- Run capillary scan — check for aggregation (spikes) and adsorption (decreasing fluorescence)
- Run MST measurement — set LED power, MST power (start at Medium), evaluation time
- Fit dose-response — calculate Fnorm, fit to 1:1 model (quadratic if [Target] > KD/10)
- Validate — run SD-test, negative control, ≥ 3 independent replicates
Buffer & Capillary Optimization
Recommended starting buffer: 50 mM HEPES pH 7.4, 150 mM NaCl, 0.05% Tween-20. Match buffer between target and ligand samples exactly.
- Tween-20/Pluronic F-127 (0.01–0.05%) — prevents adsorption
- BSA (0.05–0.5 mg/mL) — anti-adsorption, if not interfering
- DMSO — tolerated up to ~5%; keep constant across all points
- Salt — high salt (>500 mM) can reduce electrostatic MST signal
- Imidazole — keep <5–10 mM for His-tag labeling
Capillary Types
- Standard Treated — hydrophilic, general purpose (default)
- Premium Coated — enhanced hydrophilic, for sticky/low-conc proteins
- Hydrophobic — for membrane proteins in detergent
⚡ MST Power Settings
MST Power = IR laser intensity. Higher power → stronger signal but more risk of artifacts.
The Concentration Rule — MST's Design Parameter
Just as ITC has the Wiseman c-value, MST has a critical design parameter: the ratio of target concentration to KD. Getting this wrong is the #1 source of incorrect KD values.
[Target] ≤ KD/10 — Ideal
Ligand depletion is negligible. Simple binding model works. EC50 ≈ KD. This is the sweet spot.
[Target] = KD/10 to KD — Use Quadratic
Some ligand depletion. EC50 shifts right. Use the quadratic binding model and report this. KD is still reliable.
[Target] >> KD — Danger Zone
Severe ligand depletion. Apparent KD ≈ [Target]/2 regardless of actual affinity. You're measuring stoichiometry, not affinity. Reduce [Target] or report KD as an upper limit.
Data Analysis
Fnorm — The MST Observable
Fnorm = Fhot / Finitial × 1000 [‰]
Finitial = average fluorescence before IR laser ON (baseline). Fhot = average fluorescence at a defined time after IR laser ON. Two common evaluation windows:
- T-jump evaluation (~0.5–1.5 s): Captures temperature-dependent fluorescence change only (TRIC)
- MST + T-jump (~1.5–5 s): Captures thermophoretic movement + T-jump
ΔFnorm = Fnorm(bound) − Fnorm(unbound). Typical response amplitudes: 5–100‰. A ΔFnorm of 10‰ is a decent signal.
Note: Fnorm in ‰ is NanoTemper's convention. Other groups may report ΔF/F or alternative normalizations.
1:1 Binding Models
Simple (no ligand depletion): When [Target] ≪ KD:
fbound = [L] / ([L] + KD)
Quadratic (with ligand depletion): When [Target] is significant relative to KD:
fbound = {([L]+[T]+KD) − √(([L]+[T]+KD)²−4[L][T])} / (2[T])
The signal equation: Fnorm([L]) = (1−f) · Fnorm,unbound + f · Fnorm,bound
Hill & Competition Models
Hill equation (cooperative binding):
fbound = [L]n / ([L]n + KD,appn)
n > 1: positive cooperativity. n < 1: negative cooperativity or heterogeneous population. KD,app is not a true microscopic KD.
Competition assays — Cheng-Prusoff equation:
Ki = IC50 / (1 + [A] / KD,A)
Caveat: assumes competitive (same-site) binding and that [A] ≈ free [A]. Does not apply to non-competitive/allosteric displacement.
Quality Assessment Criteria
| Metric | Good | Acceptable | Poor |
|---|---|---|---|
| Signal-to-noise (ΔFnorm/σ) | >12 | 5–12 | <5 |
| Response amplitude (ΔFnorm) | >20‰ | 10–20‰ | <10‰ |
| Capillary scan shape | Flat, uniform | Minor variation | Spikes, irregular |
| Fluorescence consistency | <10% variation | 10–20% | >20% |
| KD confidence (68% CI) | <3-fold | 3–10-fold | >10-fold |
Data Interpretation — Good vs Bad MST Data
Learning to read MST data takes practice. Here are three realistic scenarios showing what to look for and how to troubleshoot.
✅ Good Data
- Traces: Smooth, reproducible across replicates. Clear T-jump + steady thermophoretic plateau. No bumps or irregularities.
- Capillary scan: Flat, uniform fluorescence. No spikes. <10% variation between capillaries.
- Dose-response: Clean sigmoidal curve. Both plateaus well-defined (unbound baseline and saturated response). ΔFnorm > 20‰, SNR > 12.
- KD: 150 ± 30 nM (68% CI within 3-fold). [Target] = 10 nM — well below KD/10. Simple model is valid.
→ Publish-ready.
⚠️ Fixable Problems
- Symptom: Initial fluorescence drops by 30% across capillaries (high [ligand] → low [ligand]). Traces look OK otherwise.
- Diagnosis: Protein adsorption at low ligand concentrations — the ligand may stabilize the protein against surface sticking.
- Fix 1: Add 0.05% Tween-20 to the buffer.
- Fix 2: Switch to premium coated capillaries.
- Fix 3: If fluorescence variation persists, add 0.5 mg/mL BSA.
→ Re-measure after buffer fix.
❌ Misleading Data
- Symptom: Beautiful sigmoidal curve, KD = 250 nM. But [Target] = 500 nM → KD ≈ [Target]/2.
- Diagnosis: Severe ligand depletion. You're measuring stoichiometry, not affinity. The real KD could be 10× lower.
- Red flag: KD always tracks [Target] — halve the protein, KD halves too.
- Fix: Reduce [Target] to ≤ KD,estimated/10. If fluorescence is too low, use the quadratic model and report the [Target] used.
→ Do NOT publish. Re-optimize.
Quality Control & Troubleshooting
🔴 Ligand-Induced Fluorescence
Symptom: Initial fluorescence varies systematically with [ligand]. Cause: Ligand quenches/enhances the dye or is itself fluorescent. Fix: Different labeling position; check ligand spectrum; run SD-test.
🔴 Buffer Mismatch
Symptom: Systematic Fnorm drift correlated with ligand dilution (not sigmoidal). Cause: Ligand in different buffer (DMSO/glycerol gradient). Fix: Ensure identical buffer in all points; run buffer-only control.
🟡 Adsorption to Capillary Walls
Symptom: Decreasing initial fluorescence at low [ligand]. Cause: Protein sticks to glass. Fix: Add 0.01–0.05% Tween-20; add BSA; use premium coated capillaries.
🟡 Aggregation
Symptom: Irregular traces, fluorescence spikes in capillary scan. Cause: Protein aggregation from heating or high concentration. Fix: Centrifuge samples; reduce MST power; more detergent.
🟡 Photobleaching
Symptom: Fluorescence decreases over time within a trace; baseline drifts down. Cause: Fluorophore degradation from excitation light. Fix: Reduce LED power; use photostable dyes (Cy5 > fluorescein).
🟡 Capillary Scan Irregularities
Symptom: Non-uniform fluorescence distribution (peaks, shoulders). Cause: Protein adsorption creating fluorescent hot spots. Fix: More detergent; premium coated capillaries; different type.
🟡 Protein Unfolding
Symptom: Irreversible fluorescence changes after IR exposure. Cause: 2–6 K increase may unfold marginally stable proteins. Fix: Lower MST power; verify stability with nanoDSF/Tycho at Tm + 6K.
🟡 Convection
Symptom: Irregular, non-reproducible trace shapes between identical samples. Cause: Convective flow in unsealed or over-heated capillaries. Fix: Ensure proper sealing; reduce MST power.
✅ The SD-Test (SDS Denaturation)
Add SDS (1–4% w/v) + DTT to the labeled protein. SDS denatures the protein → destroys the binding site. If the MST signal disappears, the signal was likely binding-specific. If it persists, it may be a dye artifact. This is a necessary but not sufficient control — run it for every new interaction.
When NOT to Use MST
MST is versatile, but it's not the right tool for every experiment. Honest assessment:
❌ When you need kinetics. MST gives only KD — no ka, kd, or residence time. Use SPR or BLI for kinetic characterization.
❌ When you need thermodynamics. MST gives no ΔH, ΔS, or direct stoichiometry. Use ITC for complete thermodynamic profiles.
❌ When binding produces no signal change. Some interactions don't alter thermophoretic properties. If ΔFnorm is negligible, try different labeling positions or switch techniques.
❌ For very weak interactions (>mM KD). MST's upper KD range is limited by maximum achievable ligand concentration (solubility). Very weak binders may not show saturation.
❌ For large kinetic screening campaigns. SPR with automated sample handling (Biacore 8K+, Carterra LSA) provides higher throughput for kinetic screening at scale.
❌ When label-free is mandatory. While UV MST exists, standard MST requires fluorescent labeling. SPR/BLI are truly label-free by default.
MST for Drug Discovery
Hit Validation & SAR
Rapid affinity ranking of compound series from HTS hits. 16-point dose-response in ~15 min. Ideal for structure-activity relationship (SAR) studies — quickly compare analogs. Low sample consumption makes it feasible for precious targets.
Fragment & Covalent Binders
Fragment screening: MST can detect fragments, though sensitivity depends on the induced signal change. Covalent binders: MST has a unique advantage — single-use capillaries mean no regeneration issues (a major SPR pain point for covalent compounds).
Ternary Complexes & Selectivity
PROTAC/molecular glue characterization: MST detects ternary complex formation in solution where SPR struggles. Selectivity profiling across multiple targets is efficient thanks to low sample requirements. Fits into biophysics cascades after primary screens and alongside SPR characterization.
Is My MST Data Publication-Ready?
Reviewers increasingly scrutinize MST data. Use this checklist before submitting.
Experimental Design
- ☐ ≥ 3 independent replicates (different days)
- ☐ Concentration range spans ≥20× above/below KD
- ☐ Negative control (non-binding molecule)
- ☐ SD-test performed
- ☐ Buffer identical across all samples
Data Quality
- ☐ SNR > 5 (ideally > 12)
- ☐ ΔFnorm > 10‰
- ☐ Initial fluorescence consistent (<20% var.)
- ☐ Capillary scan shows no aggregation
- ☐ No ligand-induced fluorescence change
Reporting
- ☐ KD ± CI (68% or 95%)
- ☐ Binding model specified (quadratic if needed)
- ☐ Fnorm evaluation window stated
- ☐ LED power & MST power reported
- ☐ Raw traces shown (not just dose-response)
- ☐ Orthogonal validation (SPR, ITC) if possible
Top 10 MST Mistakes & How to Avoid Them
MST vs SPR vs ITC — Which Technique Should I Use?
Every biophysics core facility gets this question. Use this vendor-neutral decision tree to pick the right technique for your experiment.
Do you need kinetic rate constants (ka, kd)?
→ YES: SPR or BLI (real-time binding curves)
→ NO: Do you need thermodynamic parameters (ΔH, ΔS, stoichiometry)?
→ YES: ITC (direct calorimetric measurement)
→ NO: Is your target difficult to immobilize or is surface attachment a concern?
→ YES: MST (in-solution, no surface needed)
→ NO: Is sample quantity very limited (<50 µg)?
→ YES: MST (needs ~0.5 µg per titration for a 60 kDa protein)
→ NO: Do you need to work in crude lysates or complex matrices?
→ YES: MST (His-tag labeling works directly in lysate)
→ NO: Is label-free detection mandatory?
→ YES: SPR, BLI, or ITC
→ NO: Is throughput the priority (>100 compounds)?
→ YES: SPR (Biacore 8K+) or Dianthus (384-well MST)
→ NO: MST — fast, simple, low cost per experiment
Best practice: Use MST for rapid KD screening, then confirm with SPR (kinetics) and/or ITC (thermodynamics). Orthogonal validation = high confidence.
MST vs SPR vs ITC — Complementary Techniques
🔬 MST
- In-solution: KD only (equilibrium)
- Requires fluorescent labeling
- Very low sample (~4 µL/capillary)
- KD range: sub-nM to mM
- Tolerates crude samples (lysates, serum)
- Fast (~15 min per titration)
- No kinetics, no thermodynamics
- Limited regulatory acceptance
✨ SPR / 💡 BLI
- Surface-based: ka, kd, KD (kinetics)
- Label-free detection
- Requires immobilization
- Real-time binding curves
- Extensive FDA/regulatory acceptance
- Excellent for small molecules (SPR)
- Higher throughput with multi-channel systems
🌡️ ITC
- In-solution: KD, ΔH, ΔS, n (thermodynamics)
- No labeling, no immobilization
- Higher sample consumption (~200–300 µL)
- Gold standard for thermodynamic profiling
- No kinetics (standard ITC)
- Directly measures stoichiometry
- Established regulatory acceptance
Best practice: MST is an excellent orthogonal validation tool. Combine MST (solution KD) with SPR (kinetics + surface KD) and/or ITC (thermodynamics). If KD values agree across techniques, confidence is high. MST is particularly valuable early in drug discovery for rapid screening before detailed SPR/ITC characterization. See our technique comparison page for a comprehensive overview.
Applications
Drug Discovery & Screening
Rapid affinity ranking of compound libraries. Fragment screening and hit validation. SAR studies with dose-response for each analog. Competition assays to determine binding site overlap.
Protein & Nucleic Acid Interactions
Antibody–antigen affinity (nanobodies, scFvs). Protein–DNA/RNA binding. Multiprotein complex formation. Membrane proteins in detergent micelles or nanodiscs — MST is particularly forgiving of these conditions.
Challenging & Special Samples
Cell lysate measurements with His-tag RED-tris-NTA — no purification needed. Lipid–protein interactions, metal ion binding (Ca²⁺, Zn²⁺), buffer/additive screening, and aptamer–target characterization during SELEX.
Instrumentation
All MST instruments are manufactured by NanoTemper Technologies (Munich, Germany). The platform has two form factors: capillary-based (Monolith) and plate-based (Dianthus).
Capillary-Based (Monolith Series)
- Monolith NT.115 — The workhorse. Blue (470 nm → GFP/fluorescein) and red (625–650 nm → Cy5/RED dyes) LED channels. Manual capillary loading.
- Monolith NT.115 Pico — Enhanced sensitivity for low-fluorescence applications.
- Monolith NT.Automated — Automated capillary handling from 96/384-well plates. Higher throughput for screening campaigns.
- Monolith NT.LabelFree — UV optics (280 nm) for intrinsic tryptophan fluorescence. No labeling required.
Plate-Based (Dianthus)
384-well plate format using an extrinsic dye (e.g. RED-tris-NTA / RED-NHS) with dual-wavelength detection of dye emission (e.g. ~650/670 nm for the RED dye) for spectral shift measurements. No capillaries and no temperature gradient needed.
Important: Dianthus Spectral Shift measures the ratio of dye emission at two wavelengths upon binding — this is not thermophoresis, and it is not the 330/350 nm intrinsic-tryptophan readout used by nanoDSF (Prometheus/Tycho). The physical basis is binding-induced changes to the dye's local environment that shift its emission spectrum. Higher throughput than capillary MST.
Instruments using MST
NanoTemper Monolith NT.115 — standard MST, blue/red LED · Monolith NT.115 Pico — enhanced sensitivity · Monolith NT.Automated — automated from plates · Monolith NT.LabelFree — UV/intrinsic fluorescence · NanoTemper Dianthus — plate-based, spectral shift (384-well)
Have SPR or BLI data?
Upload your raw files and get an automated kinetic analysis in minutes. We support Biacore, Octet, and other major formats.
Upload & Analyze